Staff profile
Dr Susannah Bourne-Worster
Royal Society University Research Fellow

| Affiliation | Telephone |
|---|---|
| Royal Society University Research Fellow in the Department of Chemistry |
Biography
Susannah is a computational chemist interested in understanding the photoexcited dynamics of molecules in complex environments. Having grown up a committed technophobe, Susannah was first introduced to programming and the joys of theoretical chemistry during her MChem project on ‘Localisation of Polarons in Conformationally Disordered Polymers’ under the supervision of Prof. William Barford at the University of Oxford. She remained in Oxford to complete a DPhil in Theoretical and Biophysical Chemistry with Prof. Peter Hore, studying the effect of spin relaxation in the photoinduced radical pair reaction thought to underpin the magnetic compass sense of migratory birds. In 2018, Susannah completed her DPhil, got married (changing her name from Worster to Bourne-Worster) and moved to Bristol to take up an independent postdoctoral fellowship from The Royal Commission for the Exhibition of 1851. During her fellowship, Susannah applied her expertise in Open Quantum Systems to modelling exciton transport in photosynthetic antenna and gained experience in TDDFT and semiempirical methods for excited states under the guidance of Prof. Fred Manby. In 2022, she moved to UCL to work with Prof. Graham Worth as a postdoctoral researcher on the collaborative UPDICE (Ultrafast Photo Dynamics in Complex Environments) programme between UCL, Bristol, Oxford and Imperial. During a year of maternity leave in 2024, Susannah was successful in obtaining a Royal Society University Research Fellowship to build an independent research group. She took up her fellowship in January 2025 at the University of Durham, where she also holds a proleptic post as Assistant Professor in Computational Chemistry/Data Science.
Research Interests
Overview
We are a computational group interested in exploring how the photochemistry and photophysics of light-absorbing molecules can be influenced, and controlled, through interactions with their surroundings. We are particularly interested in understanding how these interactions can be exploited in the design of more sustainable materials for solar energy.
Solar technology has improved over recent decades to the point where it is an economically viable and increasingly important part of UK and global energy strategies. However, gradual increases in efficiency have come at a cost. For example, rooftop silicon solar panels take so much energy to recycle that it is estimated that around 80 million tonnes will end up in landfill by 2050. We need to take a long-term view to designing future solar technology, considering the environmental impact of producing, using and recycling the component materials from the start.
Nature shows us how to do this. The machinery of photosynthesis uses the same non-toxic and fully recyclable components (molecules) for widely different functions, prompting a philosophy of ‘using the resources to hand’ over striving for inherently optimised but costly materials. These handy molecules are optimised for their different roles by the surrounding protein environment. Understanding this optimisation is an exciting opportunity to gain fundamental insights into light-driven chemistry and one of the most important processes of life.
By using high-quality simulations to probe the fascinating complexity of light-activated processes (like photosynthesis) in living systems, we hope to learn important lessons that can be applied to solving the global challenge of clean energy production.
Dynamical modelling of large systems
Modelling large, complex biological systems is computationally extremely challenging and a combination of techniques are required to capture effects over a wide range of time and length scales. For existing systems, a top-level, overarching model can be experimentally parameterised with descriptions of individual lower-level processes. However, for predictive design, it is essential to have ab initio evaluations of the underlying properties and dynamics. Moreover, the characteristics of new systems must be evaluated quickly and cheaply to enable exploration of a large space of possible designs.
We are interested in
1. Developing fast, accurate methods for evaluating the excited state properties of large (bio)chromophores.
2. Pushing the application of quantum dynamics methods towards larger, embedded systems.
S. Bourne Worster, O. Feighan and F. R. Manby, J. Chem. Phys. 154, 124106 (2021)
O. Feighan, F. R. Manby and S. Bourne-Worster, J. Chem. Phys. 158, 024107 (2023)
S. Bourne-Worster and G. Worth, J. Chem. Phys. 160, 065102 (2024)
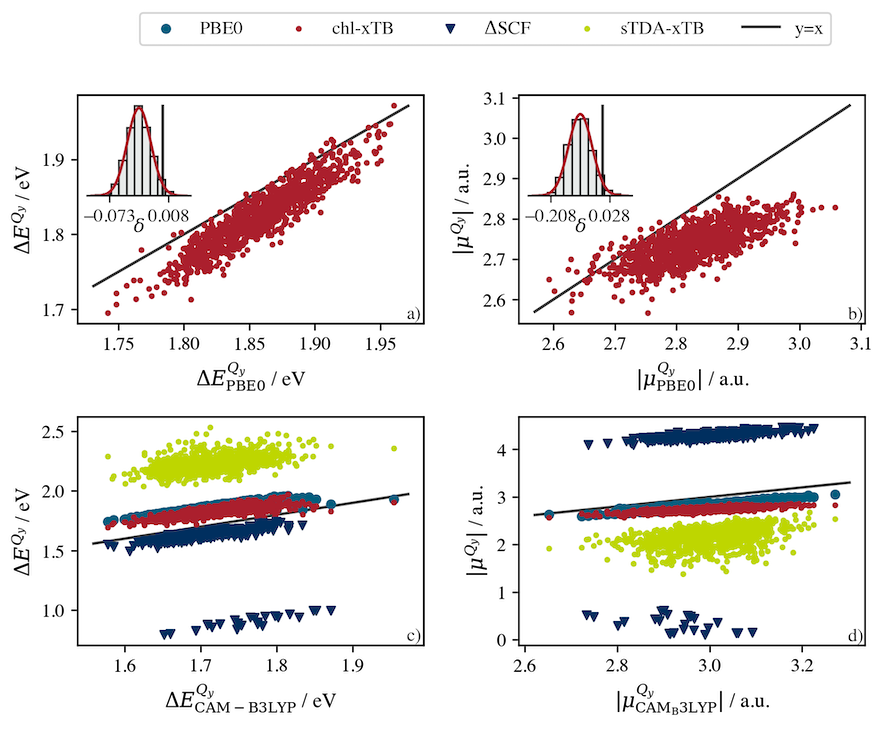
Understanding environmental control in Nature
Exciton transfer (transfer of energy between light-absorbing pigments) and charge separation (conversion of absorbed sunlight to electrons) are both fundamentally important processes in photocatalysis. Chlorophyll performs both these functions within the machinery of photosynthesis. In the antenna complexes that initially absorb sunlight, networks of chlorophyll perform highly efficient exciton transfer to transport the absorbed energy to reaction centres. In the reaction centres themselves, pairs of chlorophyll pigments undergo a symmetric charge transfer to produce the electron-hole pair that triggers the photosynthetic reaction cycle. Chlorophyll in a simple organic solvent either fluoresces or undergoes rapid non-radiative decay (at high concentrations). We are interested in characterising and comparing these different situations to isolate the features of the environment that optimise chlorophyll for its different functional roles.
S. Bourne-Worster, O. Feighan and F. R. Manby, PNAS, 120 (5) e2210811120, (2023)
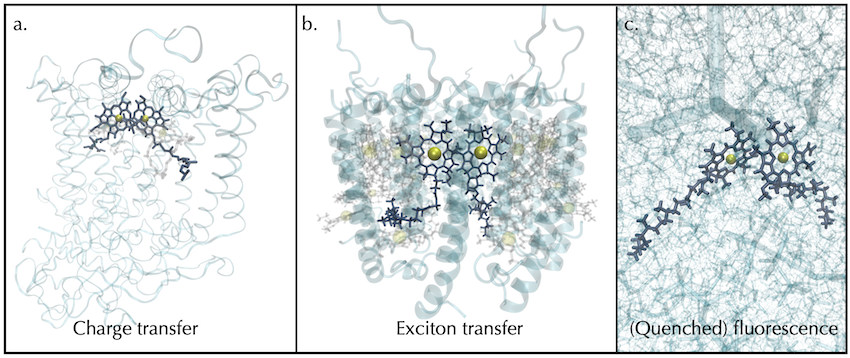
Tuneable photochemistry of organic chromophores
While chlorophyll successfully performs multiple roles in natural photosynthesis, is naturally non-toxic and biodegradable, it has a poorly-scaling synthesis, traditionally involving expensive reagents or large amounts of chlorinated solvents, making it less than ideal as the basis of a sustainable solar technology. However, the principles observed from environmentally-tuned chlorophyll photochemistry will inform promising routes to tuning the photochemistry of alternative chromophores. In the future, we will be working towards building computational workflows to generate ‘design specifications’ for supramolecular host-guest structures that could tune the behaviour of small organic chromophores for a desired functionality.
Publications
Journal Article
- Unraveling the Ultrafast Photochemical Dynamics of Nitrobenzene in Aqueous SolutionLau, N. A., Ghosh, D., Bourne-Worster, S., Kumar, R., Whitaker, W. A., Heitland, J., Davies, J. A., Karras, G., Clark, I. P., Greetham, G. M., Worth, G. A., Orr-Ewing, A. J., & Fielding, H. H. (2024). Unraveling the Ultrafast Photochemical Dynamics of Nitrobenzene in Aqueous Solution. Journal of the American Chemical Society, 146(15), 10407-10417. https://doi.org/10.1021/jacs.3c13826
- Quantum dynamics of excited state proton transfer in green fluorescent proteinBourne-Worster, S., & Worth, G. A. (2024). Quantum dynamics of excited state proton transfer in green fluorescent protein. The Journal of Chemical Physics, 160(6). https://doi.org/10.1063/5.0188834
- Charge transfer as a mechanism for chlorophyll fluorescence concentration quenchingBourne-Worster, S., Feighan, O., & Manby, F. R. (2023). Charge transfer as a mechanism for chlorophyll fluorescence concentration quenching. Proceedings of the National Academy of Sciences, 120(5). https://doi.org/10.1073/pnas.2210811120
- An efficient protocol for excited states of large biochromophoresFeighan, O., Manby, F. R., & Bourne-Worster, S. (2023). An efficient protocol for excited states of large biochromophores. The Journal of Chemical Physics, 158(2). https://doi.org/10.1063/5.0132417
- Reliable transition properties from excited-state mean-field calculationsBourne Worster, S., Feighan, O., & Manby, F. R. (2021). Reliable transition properties from excited-state mean-field calculations. The Journal of Chemical Physics, 154(12). https://doi.org/10.1063/5.0041233
- Structure and Efficiency in Bacterial Photosynthetic Light HarvestingBourne Worster, S., Stross, C., Vaughan, F. M. W. C., Linden, N., & Manby, F. R. (2019). Structure and Efficiency in Bacterial Photosynthetic Light Harvesting. The Journal of Physical Chemistry Letters, 10(23), 7383-7390. https://doi.org/10.1021/acs.jpclett.9b02625
- Proposal to use superparamagnetic nanoparticles to test the role of cryptochrome in magnetoreceptionBourne Worster, S., & Hore, P. J. (2018). Proposal to use superparamagnetic nanoparticles to test the role of cryptochrome in magnetoreception. Journal of The Royal Society Interface, 15(147), Article 20180587. https://doi.org/10.1098/rsif.2018.0587
- A light-dependent magnetoreception mechanism insensitive to light intensity and polarizationWorster, S., Mouritsen, H., & Hore, P. J. (2017). A light-dependent magnetoreception mechanism insensitive to light intensity and polarization. Journal of The Royal Society Interface, 14(134), Article 20170405. https://doi.org/10.1098/rsif.2017.0405
- Spin relaxation of radicals in cryptochrome and its role in avian magnetoreceptionWorster, S., Kattnig, D. R., & Hore, P. J. (2016). Spin relaxation of radicals in cryptochrome and its role in avian magnetoreception. The Journal of Chemical Physics, 145(3), Article 035104. https://doi.org/10.1063/1.4958624
- The quantum needle of the avian magnetic compassHiscock, H. G., Worster, S., Kattnig, D. R., Steers, C., Jin, Y., Manolopoulos, D. E., Mouritsen, H., & Hore, P. J. (2016). The quantum needle of the avian magnetic compass. Proceedings of the National Academy of Sciences, 113(17), 4634-4639. https://doi.org/10.1073/pnas.1600341113
- Three-Gene Immunohistochemical Panel Adds to Clinical Staging Algorithms to Predict Prognosis for Patients With Esophageal AdenocarcinomaOng, C. J., Shapiro, J., Nason, K. S., Davison, J. M., Liu, X., Ross-Innes, C., O’Donovan, M., Dinjens, W. N., Biermann, K., Shannon, N., Worster, S., Schulz, L. K., Luketich, J. D., Wijnhoven, B. P., Hardwick, R. H., & Fitzgerald, R. C. (2013). Three-Gene Immunohistochemical Panel Adds to Clinical Staging Algorithms to Predict Prognosis for Patients With Esophageal Adenocarcinoma. Journal of Clinical Oncology, 31(12), 1576-1582. https://doi.org/10.1200/jco.2012.45.9636